

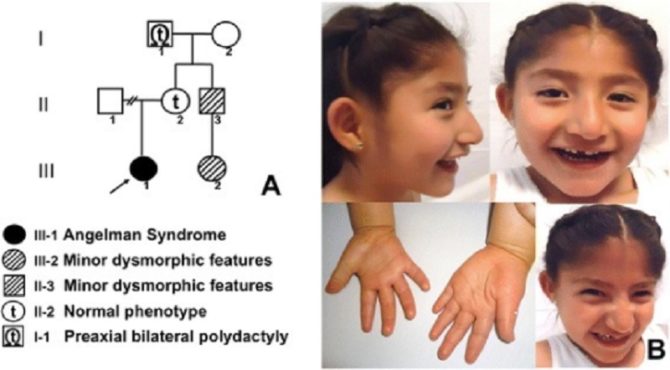
Con una incidencia de 1 en cada 15 000 a 30 000 nacidos vivos, el Síndrome de Angelman es una enfermedad genética que se caracteriza por provocar discapacidad intelectual severa. Aunque muy similar al autismo, tiene características propias que lo diferencias.
No dejes de leer sobre otra importante afección: Trastorno del espectro autista o autismo, conócelo…
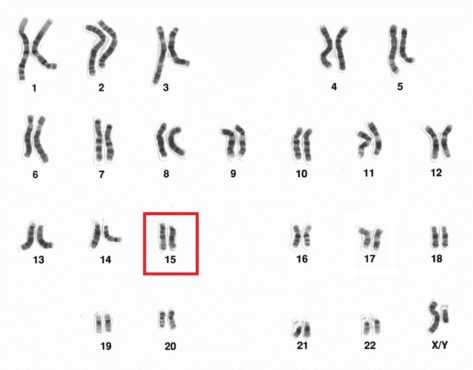
El bebé, aparentemente normal a su nacimiento, tiene una deleción en el brazo largo del cromosoma 15, pero la enfermedad no empieza a manifestarse hasta los 6 a 12 meses de edad. Los síntomas que permiten su diagnóstico claro no son evidentes hasta que el niño no alcanza los 3 a 7 años.
Síntomas del Síndrome de Angelman
Aunque hay evidencias de que esta enfermedad ha acompañado al hombre desde hace mucho, el Síndrome de Angelman no fue plenamente descrito hasta una fecha tan reciente como 1964. Lleva el nombre del pediatra Harry Angelman, que atendió a tres niños con la enfermedad, pero su base genética no se estableció hasta que en 1987 el Angelman Research Group de Florida identificó la deleción en el cromosoma 15 como causa del síndrome.
El rasgo más distintivo del síndrome de Angelman es un estado de alegría perenne en el paciente, que ríe y sonríe de forma continua, y un andar con las manos en alto. Por ello, este síndrome ha sido llamado anteriormente “de la marioneta feliz”. También son típicos los temblores y movimientos bruscos en los brazos, y piernas más rígidas de lo normal. Incide por igual a hombres y mujeres, sin distinción de sexo.

Existen otros rasgos típicos, aunque no todos tienen que estar presentes todos para que un especialista lo diagnostique:
- Habilidad para comunicarse oralmente reducida o nula.
- Baja coordinación motriz, problemas de equilibrio y movimiento.
- Ataxia.
- Poca comunicación, reducida a gestos y señales.
- Estado aparente y permanente de alegría.
- Falta de atención
- Hipermotricidad
- Retraso importante del desarrollo.
- Microcefalia.
- Crisis convulsivas (alrededor de los tres años de edad).
Síntomas más severos
El síndrome de Angelman presenta otros síntomas más severos en el 20% de los casos. Estos son:
- Achatamiento posterior de la cabeza
- Fascinación por el agua
- Babeo
- Dificultad al comer
- Estrabismo
- Epilepsia
- Hiperactividad sin agresividad
- Hipersensibilidad al calor
- Hipopigmentación en la piel
- Insomnio
- Lengua y mandíbula prominente
Sus causas genéticas son diversas, siendo las deleciones en el locus del cromosoma 15q11-q13 responsables del 75% de los casos. El 6% de los niños con discapacidad intelectual que tienen convulsiones es debido al síndrome de Angelman.
Diagnosticando el síndrome de Angelman
Un fuerte indicador que permite diferenciar a un niño con este síndrome de un niño autista es su alta sociabilidad, pues es altamente sociable, a diferencia del niño con autismo. También puede confundirse con una parálisis cerebral no específica, con el síndrome de Rett y el síndrome de Lennox-Gastaut.
Una vez sugerido el diagnóstico de síndrome de Angelman por el pediatra, se realizan pruebas genéticas para corroborar que existe una deleción en el cromosoma 15 por la prueba de FISH (Fluorescence in Situ Hybridization). Este diagnóstico puede completarse con diferentes pruebas de biología molecular y existen diversas posibilidades para llegar a un diagnostico especifico. Estos diagnosticos moleculares solo se realizan en centros muy especializados.
El mecanismo genético de aparición de la enfermedad es muy importante, pues de él varía la posibilidad de que se repita el síndrome en un próximo embarazo, con una posibilidad entre el 0 y el 50% de riesgo. En el 80% de los casos, se puede identificar el mecanismo genético y realizar un diagnóstico prenatal con células fetales producto de una biopsia corial o una amniocentesis.
Tratamiento del síndrome de Angelman
Como tal, esta enfermedad genética no tiene cura, aunque si existe tratamiento para alguno de sus síntomas. La epilepsia se trata con medicamentos anticonvulsivantes (valproato, benzodiacepinas) y se introducen en el tratamiento fármacos que induzcan el sueño, para mitigar la hiperactividad. Estos, además, ayudan a controlar los trastornos de sueño de los niños con síndrome de Angelman, que normalmente solo duermen unas 5 horas al día.
Se recomienda que los niños aquejados con esta enfermedad genética se traten con intervenciones fonoaudiológicas y kinesiológicas. También es útil recurrir a terapias de apoyo como la musicoterapia y la hidroterapia.

Se ha descrito el efecto beneficioso en estos casos de la terapia de la conducta para superar comportamientos problemáticos, la hiperactividad y el lapso atencional corto. También la logopedia ayuda a desarrollar habilidades no verbales del lenguaje, introduciendo el lenguaje de señas.
Por último, la fisioterapia les ayuda a corregir la postura y mejorar el equilibrio y la capacidad de caminar. En estos aspectos, actividades como la natación, la equitación y la musicoterapia son también beneficiosas.
Los pacientes con síndrome de Angelman tienen problemas de escoliosis frecuente, por lo que se recomienda el uso de corsés medicinales y, en los casos más severos, intervención quirúrgica para corregir las desviaciones de columna.
Pronóstico y evolución
La severidad de los síntomas de un niño afectado con el síndrome de Angelman es muy variable. Muchos desarrollan la capacidad de hablar y se comunican de forma efectiva, tienen conciencia de sí mismos y son capaces de establecer lazos afectivos con las demás personas. No obstante, sus dificultades a la hora de andar y lo básico de su lenguaje pueden provocar rechazo social.
Una atención cercana y participar en terapias ocupacionales, físicas y comunicativas de forma regular mejoran mucho el pronóstico. La enfermedad evoluciona con la edad: aunque la hiperactividad y el insomnio se incrementan, las convulsiones mejoran y hasta cesan.
Casi todos los adultos con el síndrome de Angelman pueden comer normalmente con tenedor y cuchara, y las mujeres presentan tendencia a la obesidad.
En los pacientes con síndrome de Angelman la pubertad y la adolescencia se manifiestan a edad normal y el desarrollo sexual es completo. Su salud general es buena y tienen, dentro de sus limitaciones, una esperanza de vida normal. La tendencia a la alegría constante, que es un elemento positivo en la niñez, se mantiene durante toda su vida, lo cual puede provocar rechazo social.
Aunque el síndrome de Angelman no se cura, si se trata al paciente de forma personalizada se puede mejorar significativamente su calidad de vida y favorecer su desarrollo.
Te invito a leer: Niños con necesidades especiales = demandas educativas individuales





