

También llamado trisomía 18, el síndrome de Edwards consiste en un tipo de aneuploidía humana. Cuando un cromosoma adicional se presenta en el par 18, bien sea de forma total o parcial.
Este síndrome se descubrió en el año 1960. Un especialista de la Universidad de Wisconsin, llamado John H. Edwards, encontró el fenotipo característico de la condición.
Sus resultados se registran como un descubrimiento en la literatura médica de pediatría y genética. Sin embargo, todavía no se conocen con exactitud las regiones puntuales en las que se origina.
En cuanto a genética molecular, solo se señala que hay dos regiones del brazo largo, 18q12 – 21 y 18q23, que se duplican. Acto que deriva las características físicas y fisiológicas de la afección.
Contenido
Efectos del Síndrome de Edwards
La ciencia ha podido identificar más de un centenar de defectos asociados a la trisomía 18. De ahí, que resulte necesario conocerlos para identificar su incidencia en el niño a temprana edad. Entre los síntomas de la afección en el cromosoma 18 están:
- Dimorfismos faciales.
- Deformidades músculo – esquelético.
- Malformaciones craneales.
- Anomalías faciales.
- Afecciones del cuello.
- Dificultades cardiovasculares.
- Disfunciones gastrointestinales.
- Defectos genito-uterinos.
- Singularidades en extremidades.
Dimorfismos faciales
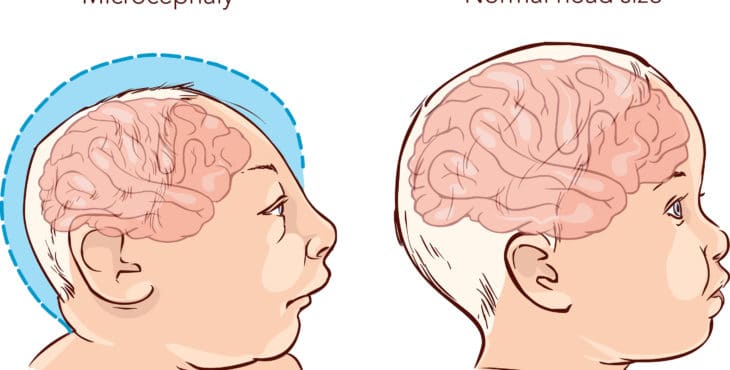
Ahora, es posible encontrar la microcefalia, el occipucio prominente, la frente estrecha y fisuras palpebrales cortas. Además, también es común que se presenten orejas malformadas, paladar ojival y micrognatia. Estos diformismos faciales se expresan en la mayoría de los casos, así que debes estar alerta a su presencia en tu pequeño.
En la microfelia, el cráneo crece hasta un tamaño menor al promedio. Ello reduce la esperanza de vida de la persona por una función cerebral pobre. En lo que respecta al occipucio prominente, se ve caracterizado por una imponente parte posterior de la cabeza.
El occipucio es coincidente con otras afecciones como orejas malformadas y micrognatia. Esta última ocurre cuando la mandíbula no se forma debidamente y es más pequeña al promedio. El paladar ojival se conoce por una mandíbula interna estrecha que induce a que se desarrollen otras afecciones como:
- Mordida cruzada lateral.
- Apiñamiento dental.
- Deficiencia de respiración nasal.
Deformidades músculo-esquelético
Pelvis angosta, cuello con exceso de piel, esternón pequeño, dislocación de caderas, focomelia, pie en mecedora, dedos sobrepuestos y uñas hipoplásicas. Esas son las deformidades músculo-esqueléticas que se relacionan directamente con el síndrome de Edwards.
Cada una representa una afección a la salud integral del paciente. Por ejemplo, el cuello alado se caracteriza por piel excedente en la zona de la nuca. Esta va desde las orejas hasta los hombros.
La focomelia es una malformación en la que no existen partes óseas o musculares en los miembros. Ocurre tanto en los superiores como en los inferiores. El bebé nace con un muñón que tiene semejanza con la aleta de una foca.
El menor de los rasgos a nivel músculo-esquelético del síndrome de Edwards son las uñas hipoplásicas. El tejido articular crece en menor largo y grosor de lo esperado, además de tener una textura diferente.
Síndrome de Edwards y las malformaciones craneales
Ventriculomegalia, mielomeningocele y quiste de fosa posterior y de plexo coroideo, son las cuatro malformaciones más comunes a nivel craneal. Todas tienen como causa la trisomía 18.
La primera consiste en la dilatación del sistema ventricular del cerebro. Este proceso puede darse por un efecto obstructivo infeccioso, inflamatorio, neoplásico o extraventricular. En todos los casos, se impide el paso del líquido cefalorraquídeo entre el tercer y cuarto ventrículo.
Su detección temprana con ecosonografía fetal permite la aplicación de un tratamiento que desacelere y evite el desarrollo. Generalmente, se recurren a métodos analgésicos, esteroideos, antibióticos o quirúrgicos después del parto.
Otra afección craneal es la mielomeningocele. Su nombre común es espina bífida. En ella, la médula de la espalda se une a los arcos vertebrales y las meninges.
Continúa:Control y seguimiento del embarazo, una garantía para ti y para tu hijo
Labio leporino
Conocido como la principal anomalía facial del síndrome de Edwards. También constituye una de las afecciones congénitas más comunes. Ocurre en 15% de los casos. Además de la separación del labio superior, la fisura labial suele estar acompañado con un paladar hendido.
Son cuatro tipos de dificultades las que tiene el labio leporino. La primera es la alimentación porque el bebé podría estar imposibilitado de succionar la leche de su mamá. Además, hay riesgo de pérdida auditiva infecciosa, dada la exposición del oído medio con la garganta.
A ellas, se les suman retrasos en el desarrollo del habla y del lenguaje, ya que la laringe presenta dificultades para emitir sonidos. Y por último, problemas dentales, en tanto los dientes no se desarrollan con normalidad.
Higroma quístico e hidrops lifagectasia
Representan dos afecciones del síndrome de Edwards. En el higroma quístico, hay presencia anormal del líquido seroso. Por su parte, en el hidrops lifagectasia existe un impedimento en las conexiones linfáticas en el área de la nuca.
Es posible que puedan confundirse entre sí. Se diferencian en la ubicación. El higroma quístico solo se encuentra detrás de la nuca. Mientras que el cuadro de hidrops, se halla en la región inguinal, del mediastino y la axila.
Dificultades cardiovasculares
Se reconocen los defectos septales, la coartación aórtica y la ductus arteriosus persistente. También es usual la transposición de grandes vasos, dextrocardia y lesiones vasculares.
En más del 80% de los casos, requiere intervención quirúrgica. En la coartación aórtica, se denota un estrechamiento de la aorta torácica que ocasiona insuficiencia cardíaca.
Por su parte, el ductus arteriosus persistente incurre en la presencia del conducto arterioso entre el sistema pulmonar y la aorta. Normalmente, solo estaría en la vida fetal. Se detecta por la existencia de un soplo y casi nunca causa problemas funcionales.
El corazón se sitúa en la parte derecha del pecho. La transposición de los grandes vasos son defectos cardíacos en los dos vasos principales. Tanto por el que entra como por el que sale sangre del corazón.
Disfunciones gastrointestinales
Para detectar estos signos del síndrome de Edwards, es necesario prestarle suficiente atención a la hernia diafragmática y la onfalocele. Adicionalmente a la hernia inguinal y el intestino ecogénico.
Una anomalía congénita en la que el diafragma tiene una abertura no esperada. Esa es la descripción de hernia diafragmática. Por su parte, el onfalocele es un defecto mayor en el que los órganos abdominales crecen fuera del cuerpo.
En lo que respecta a la hernia inguinal, es la más común en la que hay una protrusión de la cavidad abdominal. Y el intestino ecogénico o blanco es identificable por afectar el crecimiento intrauterino y ser indicador de otras anomalías congénitas.
Defectos genito-uterinos
Generalmente, la trisomía 18 desencadena problemas genito-uterinos como el riñón poliquístico, con deformidades en cuanto a estructura y función. También ocurre la hidronefrosis y la criptorquidia.
El riñón poliquístico se expresa por la formación anormal de quistes en la zona renal. Tiende a afectar la función del hígado y el páncreas. Muy raramente incide en el corazón y el cerebro.
En cuanto a la criptorquidia, es aquel trastorno en el que no descienden uno o ambos testículos. Puede ser una afección hereditaria. El diagnóstico temprano asegura el mejor de los pronósticos con un tratamiento temprano.
Además, disminuye los riesgos de padecer efectos contraproducentes que desmejoren la calidad de vida. Los tratamientos pueden ser hormonales o quirúrgicos.
Singularidades en las extremidades
Ante una anomalía del cromosoma 18, las extremidades suelen verse afectadas. En relación con el Síndrome de Edwards, se identifican solo 3 hasta ahora. Estas son la polidactilia, el pie bot y la clinodactilia.
La primera es un trastorno genético en el que el bebé nace con más dedos en la mano o en el pie. Lamentablemente, solo es detectable una vez dado el nacimiento. Esos dedos extra son llamados supernumerarios.
Conocido también como pie zambo o equinovaro, el pie bot se caracteriza porque el miembro se torna hacia adentro. No tiene tratamiento por lo que el paciente se ve en la necesidad de caminar apoyándose en sus tobillos. Ocurre en 1 de cada 1000 neonatos.

Síndrome de Edwards, ¿qué debes esperar?
No se puede dejar a un lado que mueren muchos bebés con trisomía 18. Esto hace que no exista un tratamiento eficaz cuando es diagnosticada.
En todo caso, el médico alerta a la familia acerca de la presencia del síndrome de Edwards y sus implicaciones. Se requiere preparación psicológica para afrontar cualquiera de las anomalías que deriva, incluyendo, la muerte perinatal.
Usualmente, el examen de diagnóstico se realiza entre la semana 12 y la 20. El procedimiento consiste en realizar una amniocentesis o biopsia placentaria para confirmar o descartar la condición. Con la aplicación de técnicas ultrasonográficas, es posible prever una malformación.
La mayoría de los diagnósticos no llega a término. Hay más riesgo conforme la madre aumenta de edad, especialmente, si ya ha cumplido los 32 años.





